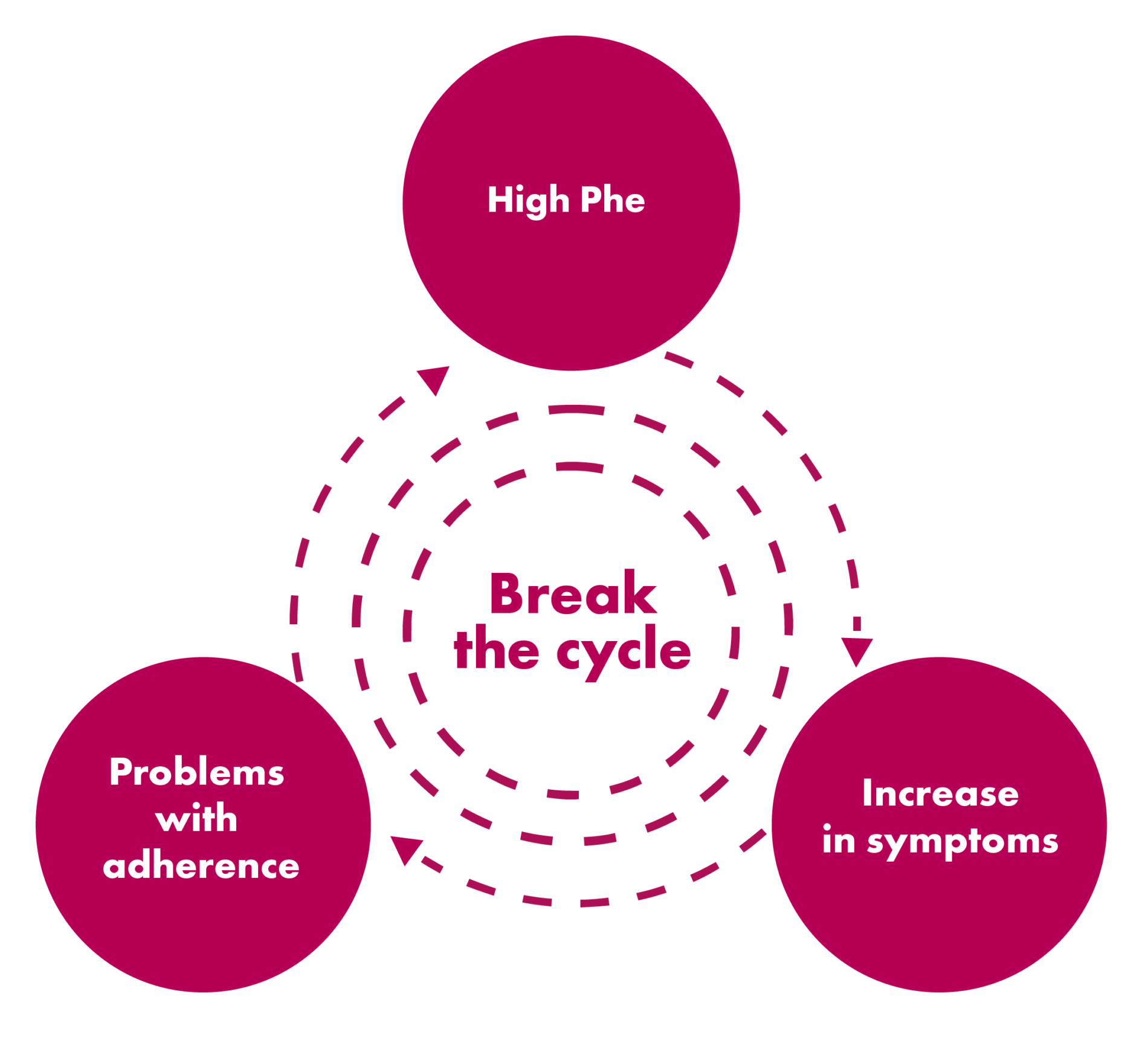
Guidelines recommend lifetime maintenance of Phe concentrations ≤360 μmol/L. However, Phe control typically drops off during adolescence, increasing the severity of neuropsychological symptoms and negatively impacting life trajectory 2,3



Adapted from Jurecki et al. Mol Genet Metab. 2017.
Compliance with dietary management, regular monitoring of blood Phe levels, clinic appointments, and the mental and emotional stress of living with a chronic disorder can make maintaining blood Phe levels within the recommended range more difficult for patients over time. Helping patients and caregivers talk openly about their challenges living with PKU may keep them engaged.4-7

Kelly: Loss of Phe control
Learn how a strong support system helped Kelly return to clinic
References: 1. Jurecki ER, Cederbaum S, Kopesky J, et al. Adherence to clinic recommendations among patients with phenylketonuria in the United States. Mol Genet Metab. 2017;120(3):190-197. 2. Brown CS, Lichter-Konecki U. Phenylketonuria (PKU): a problem solved? Mol Genet Metab Rep. 2016;6:8-12. 3. Vockley J, Andersson HC, Antshel KM, et al. American College of Medical Genetics and Genomics Therapeutic Committee. Phenylalanine hydroxylase deficiency: diagnosis and management guidelines. Genet Med. 2014;16(2):188-200. 4. Enns GM, Koch R, Brumm V, Blakely E, Suter R, Jurecki E. Suboptimal outcomes in patients with PKU treated early with diet alone: revisiting the evidence. Mol Genet Metab. 2010;101(2-3):99-109. 5. van Wegberg AMJ, MacDonald A, Ahring K, et al. The complete European guidelines of phenylketonuria: diagnosis and treatment. Orphanet J Rare Dis. 2017;12(1):162. doi: 10.1186/s13023-017-0685-22. 6. Cazzorla C, Bensi G, Biasucci G, et al. Living with phenylketonuria in adulthood: the PKU ATTITUDE study. Mol Gen Metab Rep. 2018;16:39-45. 7. Thomas J, Nguyen-Driver M, Bausell H, Breck J, Zambrano J, Birardi V. Strategies for successful long-term engagement of adults with phenylalanine hydroxylase deficiency returning to the clinic. J Inborn Errors Metab Screen. 2017;5:1-9. 8. Bilder DA, Noel JK, Baker ER, et al. Systematic review and meta-analysis of neuropsychiatric symptoms and executive functioning in adults with phenylketonuria. Dev Neuropsychol. 2016;41(4):245-260.