To date, 11 different enzymatic deficiencies are known to cause seven MPS disorders:2,4
MPS disorders exhibit similar pathologies and mechanisms, regardless of the specific enzyme deficiency:1,6
As an example, this video illustrates the mechanism of disease for Morquio A (MPS IVA).
To date, 11 different enzymatic deficiencies are known to cause seven MPS disorders:2,4
Many patients with mucopolysaccharidosis (MPS) disorders experience significant diagnostic delay because of disease rarity, phenotypic heterogeneity, and a myriad of seemingly unrelated symptoms associated with the disease.1,7,8 In a survey of patients with rare diseases (N=920) sponsored by the Global Genes Project, the mean reported length of time from symptom onset to accurate diagnosis was 4.8 years, with a range of 0 to 20 years.9
The emergence of specific therapies for some MPS disorders has increased the need for accurate and early diagnosis.1,11,12-14 Any delay in diagnosis or treatment may result in:
Since treatment exists for a number of MPS disorders, delay in diagnosis can mean delay in therapy and/or delay in disease-specific management.3,13,14
MPS disorders are clinically heterogeneous and result in serious complications regardless of the rate of progression, which can range from slowly to rapidly progressing. While rates of progression are gross characterisations of disease severity, patients with slowly progressing disease are also at risk for the serious morbidities and mortality commonly associated with rapidly progressing disease.7,3,18

Images depict non-classical MPS patient (top) and classical MPS patient (bottom)
Irrespective of phenotype, symptoms of MPS can progress into end-organ damage16
The best way to accurately diagnose MPS is to refer a patient to a geneticist who is familiar with MPS and knows how to confirm the diagnosis.1,22
You should suspect MPS when confronted by patients who:
When you suspect MPS, refer immediately to a geneticist or metabolic centre.1,22 Read more about testing and assessment.
Early identification relies on seeing the full clinical picture and knowing the key clinical hallmarks of MPS.1,3,7,14,22
An optimal care-delivery model for MPS disorders
Decades of disease and therapeutic research and practice have culminated in new approaches to clinical management of MPS disorders. At the centre of today’s new era of management is – a coordinated, multidisciplinary care-delivery model.3,23-24
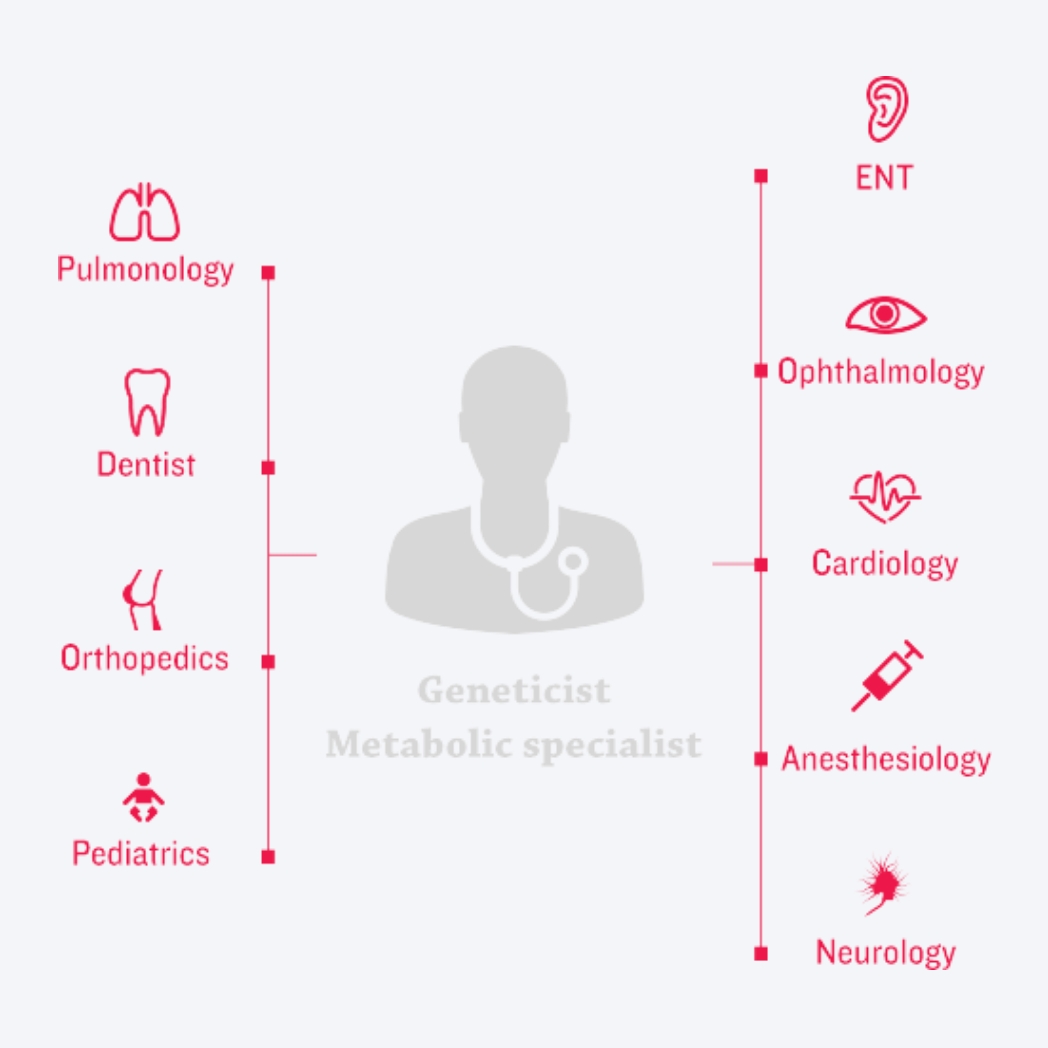
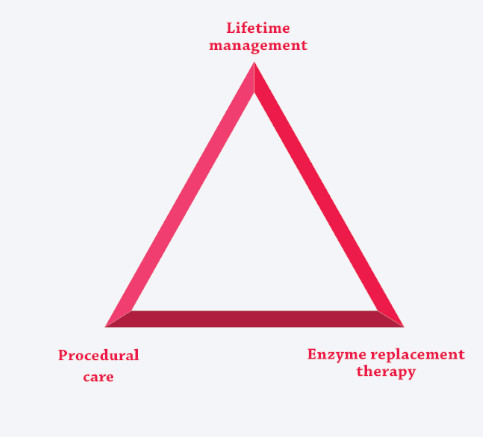
The unique risks and needs associated with these multisystemic, complex conditions are best addressed by coordinating ERT (if available) with the care-delivery model, creating three pillars of care.3,7,12,25-27
As the management paradigm continues to evolve, changes in best practices and strategies continue to show greater promise for the patients and families affected by the disorders.12,23
Prompt referral for diagnosis is essential to optimise care for patients with MPS.1,3,7
Get access to the latest webinars, meetings and events about MPS; registration is quick and easy.