While each subtype of MPS disorder is clinically distinct, all feature the life-limiting, progressive, multisystemic disease manifestations common to MPS disease pathology.2,3,4,5 Management of patients with MPS requires an understanding of the specific clinical manifestations and management recommendations for each MPS subtype.2,6
Disease name: Hurler, Hurler/Scheie, Scheie | Deficient enzyme: α-L-iduronidase | Gene symbol: IDUA
MPS I is a progressive condition2 that has been divided into 3 subtypes known as Hurler syndrome (MPS I-H), Hurler-Scheie syndrome (MPS I-H/S), and Scheie syndrome (MPS I-S).3 All subtypes of MPS I are caused by a deficiency of the enzyme α-L-iduronidase, which is required for the degradation of the glycosaminoglycans (GAGs) heparan sulphate and dermatan sulphate,2,4 with resulting progressive, multisystemic manifestations.2
1. Beck M et al. The natural history of MPS I: global perspectives from the MPS I Registry. Genet Med.2014;16(10):759–765.
2. Clarke LA et al. Long-term efficacy and safety of laronidase in the treatment of mucopolysaccharidosis I. Pediatrics 2009;132(1):229–240.
3. Yasuda E et al. Long-term follow-up of post hematopoietic stem cell transplantation for Hurler syndrome: clinical, biochemical, and pathological improvements. Mol Genet Metab Rep 2015;2:65–76.
4. Shapiro EG et al. Neurocognition across the spectrum of mucopolysaccharidosis type I: age, severity, and treatment. Mol Genet Metab 2015;116(1-2):61–68.
5. Muenzer J et al. International Consensus Panel on the Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics 2009;123(1):19–29.
6. Arn P et al. Characterization of surgical procedures in patients with mucopolysaccharidosis type I: findings from the MPS I Registry. J Pediatr 2009;154(6):859–864.e3.
7. Thomas JA et al. Childhood onset of Scheie syndrome, the attenuated form of mucopolysaccharidosis I. J Inherit Metab Dis 2010;33(4):421–427.
8. Semenza GL et al. Respiratory complications of mucopolysaccharide storage disorders. Medicine 1988;67(4):209–219.
9. Clarke LA et al. Mucopolysaccharidosis type I. In: Pagon RA et al eds. GeneReviews® Seattle, WA: University of Washington, Seattle; 2002. http://www.ncbi.nlm.nih.gov/books/NBK1162/?report=reader. Accessed June, 2023.
10. de Ru MH et al. Enzyme replacement therapy and/or hematopoietic stem cell transplantation at diagnosis in patients with mucopolysaccharidosis type I: results of a European consensus procedure. Orphanet J Rare Dis. 2011;6(55):1–9.
Disease name: Hunter | Deficient enzyme: Iduronate 2-sulphatase | Gene symbol: IDS
MPS II, also known as Hunter syndrome, is caused by a genetic pathogenic variant in the iduronate 2-sulphatase (IDS) gene leading to deficient cleavage of glycosaminoglycans (GAGs), heparan and dermatan sulphate, which leads to intracellular progressive GAG accumulation with resulting progressive, multisystemic disease.1,2
1. Hopwood JJ et al. Molecular basis of mucopolysaccharidosis type II: mutations in the iduronate 2-sulphatase gene. Hum Mutat 1993;2(6):435–442.
2. Wraith JE et al. Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr 2008;167(3):267–277.
3. Scarpa M et al. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J Rare Dis 2011;6:72.
4. Baehner F et al. Cumulative incidence rates of the mucopolysaccharidoses in Germany. J Inherit Metab Dis 2005;28(6):1011–1017.
5. Guffon N et al. Diagnosis, quality of life, and treatment of patients with Hunter syndrome in the French healthcare system: a retrospective observational study. Orphanet J Rare Dis 2015; 10:43.
6. Chkioua L et al. Molecular analysis of iduronate -2- sulfatase gene in Tunisian patients with mucopolysaccharidosis type II. Diagn Pathol 2011;6:42.
Disease name: Sanfilippo [A, B, C, D] | Deficient enzymes: Heparan N-sulphatase, α-N-acetylglucosaminidase, Acetyl CoA: α-glucosaminide N-acetyltransferase, N-acetylglucosamine-6-sulphatase | Gene symbol: SGSH, NAGLU, HGSNAT, GNS
MPS III, also known as Sanfilippo syndrome, is caused by a deficiency of 1 of 4 enzymes—heparan N-sulphatase, α-N-acetylglucosaminidase, acetyl CoA:α-glucosaminide N-acetyltransferase, and N-acetylglucosamine-6-sulphatase—which correlate with the disease’s 4 subtypes, MPS IIIA, IIIB, IIIC, and IIID, respectively. The resulting intracellular accumulation of the glycosaminoglycan (GAG) heparan sulphate leads to progressive multisystemic disease, of which central nervous system degeneration is a hallmark feature.1,2
1. Andrade F et al. Sanfilippo syndrome: overall review. Pediatr Int 2015;57(3):331–338.
2. Yogalingam G et al. Molecular genetics of mucopolysaccharidosis type IIIA and IIIB: diagnostic, clinical, and biological implications. Hum Mutat 2001;18(4):264–281.
3. Baehner F et al. Cumulative incidence rates of the mucopolysaccharidoses in Germany. J Inherit Metab Dis 2005;28(6):1011–1017.
4. Grant S et al. Parental social support, coping strategies, resilience factors, stress, anxiety and depression levels in parents of children with MPS III (Sanfilippo syndrome) or children with intellectual disabilities (ID). J Inherit Metab Dis 2013;36(2):281–291.
Disease name: Morquio A, Morquio B | Deficient enzymes: N-acetylgalactosamine 6-sulphatase, β-galactosidase| Gene symbol: GALNS, GLBI
Mucopolysaccharidosis Type IV (MPS IV), or Morquio syndrome, is a progressive, multisystemic lysosomal storage disorder resulting from a deficiency of the enzymes N-acetylgalactosamine 6-sulphatase or β-galactosidase that are responsible for the catabolism of glycosaminoglycans (GAGs),2 which are involved in the building of bones, cartilage, skin, tendons, and many other tissues in the body.3 Two distinct forms are recognised: type A (MPS IVA, Morquio A) and type B (MPS IVB). These types differ in the genetic cause of disease, but signs and symptoms among these forms can be similar and present across the spectrum of disease progression.4
MPS IV is progressive, systemic and lifelong, and it can lead to systemic morbidities and a shortened life span.1
Perceived disease rarity, heterogeneous presentation, and variability in disease progression make diagnosis challenging and early intervention critical.1 Regardless of phenotype, symptoms can progress into end organ damage.
Patients with MPS IV, which is commonly perceived as a musculoskeletal condition,1 can present with or develop unpredictable and clinically heterogeneous symptomatology extending well beyond the obvious manifestations.6 The table below illustrates the potential signs and symptoms of Morquio A, which may be observed either alone or in combination with others, and should raise your suspicion of Morquio A.
Download the Clinical Manifestations of Morquio A
Progressive, systemic manifestations can lead to potentially severe cardiovascular,1,7–9 pulmonary1,6,7,10,11 neurological,6,12 musculoskeletal,1,6 rheumatological,5,13 ophthalmological1,14,15 ENT1,16 hepatic/abdominal,13 and dental6,17 consequences. In contrast with other MPS disorders, however, patients with MPS IV do not present with cognitive impairment.
Of note, over 70% of patients manifest with unusual skeletal features within the first 2 to 3 years of life.6 Recent research has indicated that approximately 25% of patients with Morquio A syndrome present with a non-classical phenotype.5
Risk factors for increased morbidity include:4,5
The majority of patients with Morquio A do not survive past the second decade of life, with frequent causes of death including respiratory failure, complications from surgery, and cardiac failure.1
Patients can present with classical or non-classical patterns of signs and symptoms. Patients with non-classical phenotype are often reported to have significantly delayed time to diagnosis relative to symptom manifestation.1
Variable disease progression in patients with Morquio A18

Regardless of phenotype,1 symptoms can progress into end-stage organ damage.
Underappreciation of non-classical presentation in Morquio A can lead to delayed or missed diagnoses, with significant impacts:
Surgical considerations
Surgical need and burden is high among patients with Morquio A. According to a natural history study of Morquio A, 70% of the population (mean age 14.5 years) had at least 1 surgical procedure.7
Incidence of common surgical procedures for Morquio A population7,a

Adapted from Harmatz, Mol Genet Metlab 2013
Data based on medical history reviews of 325 patients with Morquio A and a mean age of 14.5 years.
A patient’s surgical history and need should alert you to the possibility of MPS IV.
MPS IVA (Morquio A) and MPS IVB are caused by pathogenic variants in the GALNS and GLB1 genes, which encode the enzymes N-acetylgalactosamine 6-sulphatase and β-galactosidase, respectively.1,25 The resulting enzyme deficiencies lead to multiple metabolic pathologies including, most notably, the accumulation of the GAG substrates keratan sulphate and chondroitin-6-sulphate in lysosomes throughout the body.1,26
Over 220 pathogenic variants identified to date in the GALNS gene give rise to wide genotypic and phenotypic heterogeneity.1
As lysosomes accumulate, they occupy an increasingly greater area of the cytoplasm, obscuring other organelles and disrupting function.27 The defective enzyme activity in MPS IV leads to cell, tissue, and organ system dysfunction that results in the progressive multisystemic morbidities that are the hallmark of this disorder.1,6

Ongoing research is transforming patient management:
Enzyme Replacement Therapy (ERT)
Published in 2014, the “International Guidelines for the Management and Treatment of Morquio A Syndrome” establish the standard of care for Morquio A. Due to the progressive nature of the disease, these guidelines urge early initiation of treatment with ERT.
In addition to ERT, ongoing lifetime management and procedural care from a multidisciplinary coordinated care team are critical components to optimising patient outcomes.6 Supportive care includes both medications and surgical interventions, including the following:1,6,7
Due to the unpredictable, multisystemic nature of MPS IVA, regular, multidisciplinary, comprehensive evaluation and treatment from a coordinated care team is also essential to identify signs of organ damage and ensure optimal patient outcomes.1,6
Physicians can optimise their management by creating a personalised management plan for each of their patients, starting with a thorough assessment schedule for each body system affected by MPS IVA.1
Download On-going multisystemic assessments in patients with Morquio A
Once diagnosis is confirmed, baseline assessments should be performed and ERT should be promptly initiated. ERT can significantly improve endurance, as measured by the 6-Minute Walk Test (6MWT). Significant change in the 6MWT reflects improvement or decline in the functional status of the cardiac, respiratory, and musculoskeletal systems and a change in disease progression.1,26 Beyond ERT, ongoing comprehensive assessments, symptomatic treatment, and surgical interventions,1,7 continuity of care from the paediatric to the adult setting is a critical consideration for patients and families living with MPS.1,30
Patients with MPS IV often require surgical intervention to address the multisystemic complications of the disease.7 This surgical care is complicated by the nature of the disease.
Patients with MPS IV suffer from multiple factors that can dramatically increase surgical risk and the need for monitoring:31
These factors complicate surgical and anesthetic care, require preplanning, and necessitate disease-specific techniques to increase optimal outcomes.31
The benefits of a procedure in patients with MPS IV should always be balanced against the associated risks.31
Specialized perioperative procedures during anesthesia, such as intubation and extubation, and the use of an intraoperative neuromonitoring checklist, are essential to successful surgical interventions.1,31 An integrated surgical team consisting of MPS VI specialists is crucial for positive, durable outcomes.1
As patients with MPS IV reach adulthood, their relationship with their medical team will change. To help manage this transition, individual plans are necessary to minimise treatment interruptions, extend support beyond the scope of paediatric care and parental support, and ensure that adult patients are knowledgeable in managing MPS IV.1,30
These transition plans should be tailored to each patients specific needs, so that those who can take over their own care will have the necessary tools, and those who are more limited will have the appropriate care and services in place to support them. The plans should include an assessment to determine the patients capacity to successfully achieve his or her outlined goals, as well as his or her knowledge and ability to communicate information about their condition.30
1. Hendriksz CJ et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A 2014;9999A:1–15.
2. Northover H et al. Mucopolysaccharidosis type IVA (Morquio syndrome): a clinical review. J Inherit Metab Dis 1996;19(3):357–365.
3. Islam T et al. Chemistry, biochemistry, and pharmaceutical potentials of glycosaminoglycans and related saccharides. In: Wong C-H, ed. Carbohydrate-based Drug Discovery Weinheim, Germany: WILEY-VCH Verlag GmbH & Co KGaA; 2003:407–439.
4. Tomatsu S et al. Mutation and polymorphism spectrum of the GALNS gene in mucopolysaccharidosis IVA (Morquio A). Hum Mutat 2005;26(6):500–512.
5. Montaño AM et al. International Morquio A Registry: clinical manifestation and natural course of Morquio A disease. J Inherit Metab Dis 2007;30(2):165-174.
6. Tomatsu S et al. Mucopolysaccharidosis type IVA (Morquio A disease): clinical review and current treatment: a special review. Curr Pharm Biotechnol 2011;12(6):931–945.
7. Harmatz P et al. The Morquio A clinical assessment program: baseline results illustrating progressive, multisystemic clinical impairments in Morquio A subjects. Mol Genet Metab 2013;109(1):54–61.
8. John RM et al. Echocardiographic abnormalities in type IV mucopolysaccharidosis. Arch Dis Child 1990;65(7):746–749. 9. Ireland MA, Rowlands DB. Mucopolysaccharidosis type IV as a cause of mitral stenosis in an adult. Br Heart J 1981;46(1):113–115.
10. Semenza GL, Pyeritz RE. Respiratory complications of mucopolysaccharide storage disorders. Medicine 1988;67(4):209–219.
11. Pelley CJ et al. Tracheomalacia in an adult with respiratory failure and Morquio syndrome. Respir Care 2007;52(3):278–282.
12. Gulati MS et al. Morquio syndrome: a rehabilitation perspective. J Spinal Cord Med 1996;19(1):12–16.
13. Holzgreve W et al. Morquio syndrome: clinical findings in 11 patients with MPS IVA and 2 patients MPS IVB. Hum Genet 1981;57(4):360–365.
14. Danes BS. Corneal clouding in the genetic mucopolysaccharidoses: a cell culture study. Clin Genet 1973;4(1):1–7.
15. Leslie T et al. Morquio syndrome: electron microscopic findings [letter]. Br J Ophthalmol 2005;89(7):917–929.
16. Hendriksz CJ et al. Review of clinical presentation and diagnosis of mucopolysaccharidosis IVA. Mol Genet Metab 2013;110:54–64.
17. Kinirons MJ, Nelson J. Dental findings in mucopolysaccharidosis type IV A (Morquio’s disease type A). Oral Surg Oral Med Oral Pathol 1990;70(2):176–179.
18. Data on file. BioMarin Pharmaceutical Inc.
19. Berger KI et al. Respiratory and sleep disorders in mucopolysaccharidosis. J Inherit Metab Dis 2013;36(2):201–210.
20. Hendriksz C. Improved diagnostic procedures in attenuated mucopolysaccharidosis. Br J Hosp Med 2011;72(2):91–95.
21. Clarke LA. Pathogenesis of skeletal and connective tissue involvement in the mucopolysaccharidoses: glycosaminoglycan storage is merely the instigator. Rheumatology (Oxford) 2011;50(suppl 5):v13–18.
22. Lehman TJA et al. Diagnosis of the mucopolysaccharidoses. Rheumatology 2011;50(suppl 5):v41–v48.
23. Morishita K, Petty RE. Musculoskeletal manifestations of mucopolysaccharidoses. Rheumatology 2011;50(suppl 5):v19–v25.
24. Muenzer J. Overview of the mucopolysaccharidoses. Rheumatology (Oxford). 2011;50(suppl 5):v4–v12.
25. Wood TC et al. Diagnosing mucopolysaccharidosis IVA. J Inherit Metab Dis 2013;36(2):293–307.
26. VIMIZIM [package insert]. Novato, CA: BioMarin Pharmaceutical Inc; 2014.
27. Coutinho MF et al. Glycosaminoglycan storage disorders: a review. Biochem Res Int 2012;2012:471325.
28. Bank RA et al. Deficiency in N-acetylgalactosamine-6-sulfate sulfatase results in collagen perturbations in cartilage of Morquio syndrome A patients. Mol Genet Metab 2009;97(3):196–201.
29. Harmatz P et al. Longitudinal analysis of endurance and respiratory function from a natural history study of Morquio A syndrome. Mol Genet Metab 2015;114(2):186–194.
30. American Academy of Pediatrics, American Academy of Family Physicians, American College of Physicians, Transitions Clinical Report Authoring Group, Cooley WC, Sagerman PJ. Supporting the health care transition from adolescence to adulthood in the medical home. Pediatrics 2011;128(1):182–200.
31. Theroux MC et al. Anesthetic care and perioperative complications of children with Morquio syndrome. Paediatr Anaesth 2012;22(9):901–907.
Disease name: Maroteaux-Lamy | Deficient enzymes: N-acetylgalactosamine 4-sulphatase | Gene symbol: ARSB
Mucopolysaccharidosis Type VI (MPS VI), or Maroteaux-Lamy syndrome, is a devastating, progressive and heterogeneous disorder with severe pathologies affecting multiple organs. It is caused by deficient activity of N-acetylgalactosamine-4-sulphatase (also known as arylsulphatase B [ASB]), the enzyme that catabolises the glycosaminoglycans (GAGs) dermatan sulphate and chondroitin sulphate.1,2
Although there is wide variability in the phenotypic presentation of MPS VI,1,2 patients are often characterised as having either rapidly or slowly progressing disease:
Regardless of rate of progression, untreated MPS VI can progress over years and lead to:3
MPS VI is a clinically heterogeneous condition in which patients can present with marked disease in the first year of life or with disease that progresses more slowly, with symptoms presenting over a longer period of time. However, it is important to recognise that MPS VI manifests symptoms along a continuum, so there are no fixed parameters for these categories of disease progression.1,2
Rapidly progressing MPS VI, characterised in the cross-sectional survey study by Swiedler et al as urinary levels of GAG protein (uGAG) above 200 μg/mg,3 can manifest in the first year of life with nonspecific symptoms. Between years 2 and 3, characteristic features develop, including:1,2,5
Slowly progressing MPS VI, characterised as uGAG levels less than or equal to 200 μg/mg, may not overtly present, thus delaying diagnosis until later in life. Nevertheless, patients develop significant morbidity that may be life limiting and life threatening. Whether rapidly or slowly progressing, MPS VI does not typically cause neurocognitive deficits, although physical limitations can affect patients’ learning and development.3
The table below outlines complications of MPS VI by organ system. The clinical manifestations associated with MPS VI are heterogeneous; however, all patients will experience disease progression.
Download Clinical Manifestations of MPS VI
Patients with MPS VI express markedly lower growth rates than unaffected age-adjusted peers. The deviation in growth experienced by patients with MPS VI may be a sign of secondary problems and should prompt clinical surveillance for issues such as malnutrition, endocrine abnormalities or psychosocial deprivation.
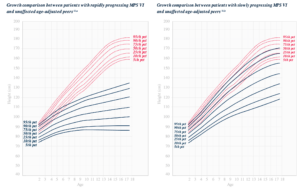
MPS VI is a clinically heterogeneous condition with a spectrum of phenotypes that have variable onset and rates of progression.2 The figure below demonstrates the wide variability between a rapidly progressing patient and a slowly progressing patient. The rapidly progressing patient displays many of the phenotypic hallmarks of MPS VI.
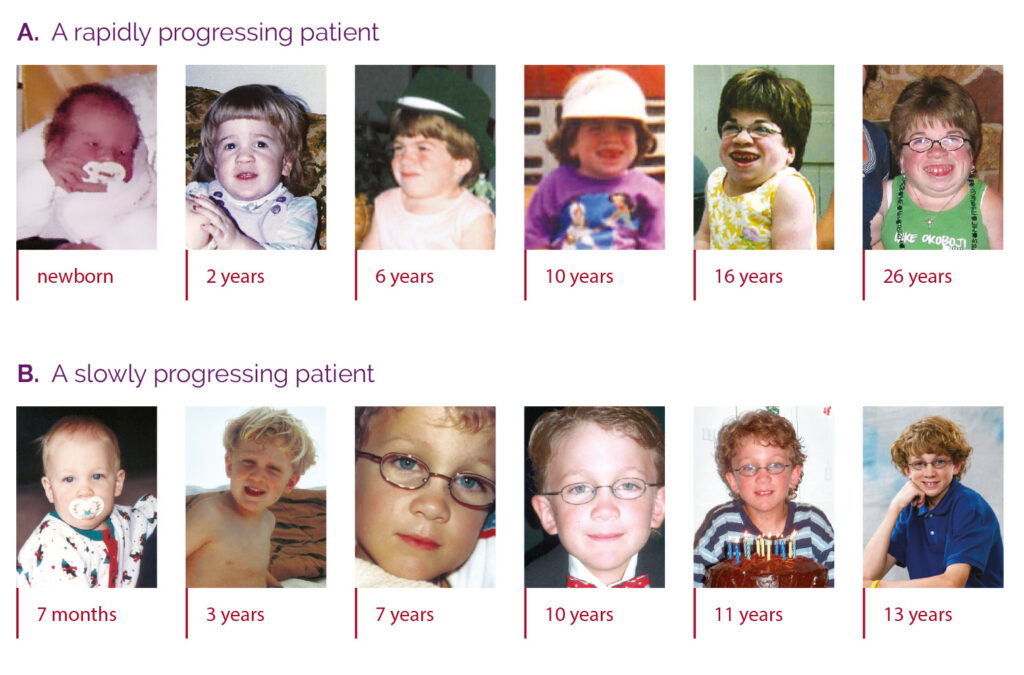
Differentiation into rapidly and slowly progressing disease is only apparent in extreme cases. If left untreated, disease progression will inhibit physical and functional well-being and results in a markedly shortened life span.4
Regardless of phenotype, affected individuals progress over the course of years and ultimately experience:4
Patients with MPS VI usually die from infections, complications secondary to surgery or cardiopulmonary disease.4
MPS VI is an autosomal recessive condition caused by a deficiency of the enzyme N-acetylgalactosamine 4-sulphatase (also known as ASB). This deficiency results in the accumulation of the GAG dermatan sulphate in the lysosomes of a wide range of tissues.13,14
Clinical manifestations of MPS VI are related to progressive accumulation of dermatan sulphate and sulphated oligosaccharides derived from both dermatan sulphate and chondroitin sulphate in lysosomes, cells and tissues.2
No specific ethnic group has been associated with an increased risk of MPS VI; however, some increased frequencies of specific pathogenic variants have been reported. There is a common pathogenic variants (1533del23) found in 23% of alleles in Brazilian patients with MPS VI. Additionally, one study demonstrated a high birth prevalence of MPS VI in the Turkish population living in Germany, as compared with the non-Turkish German population.2
Over the past decade, the management of MPS VI has evolved along with clinicians’ knowledge about the disease, such as its phenotypic variability and progression. Other factors, such as patients living into adulthood, highlight new areas for research, understanding and promise in management.
MPS VI affects multiple body systems, making management of a diverse spectrum of disease manifestations an important part of providing integrated care. Management should include use of adaptive or supportive devices, physical and occupational therapy, symptom-based medications, surgical interventions and treatment to provide the deficient enzyme.4
Published in 2007, the “Management Guidelines for Mucopolysaccharidosis” provides recommendations across specialties for the management of patients with MPS VI. It recommends enzyme replacement therapy (ERT) as a treatment option to be considered in patients with MPS VI.4
In addition to initiating ERT where appropriate, the guidelines recommend system-specific management strategies for symptoms associated with GAG buildup.4
Lifetime management and procedural care from a multidisciplinary coordinated care team are critical components to optimising patient outcomes.14,15 Patients with MPS VI require early and regular assessments to evaluate disease progression across multiple organ systems and to detect and treat potential complications of the disease.4
Download the schedule of assessments for patients with MPS VI recommended by the 2007 guidelines.
Patients with MPS VI often require surgical intervention to address the multisystemic complications of the disease. This surgical care is complicated by the nature of the disease.15
Patients with MPS VI suffer from multiple factors that can dramatically increase surgical risk and the need for monitoring, including:15
These factors complicate surgical and anaesthetic care, require preplanning and necessitate disease-specific techniques to increase optimal outcomes.16
The benefits of a procedure in patients with MPS VI should always be balanced against the associated risks.15
Specialised perioperative procedures during anaesthesia, such as intubation and extubation, and use of an intraoperative neuromonitoring checklist are essential to successful surgical interventions. An integrated surgical team consisting of MPS VI specialists is critical for positive, durable outcomes.15,16
As patients with MPS VI reach adulthood, their relationship with their medical team will change. To help manage this transition, individual plans are necessary to minimise treatment interruptions, extend support beyond the scope of paediatric care and parental support, and ensure that adult patients are knowledgeable in managing MPS VI.17
These transition plans should be tailored to each patient’s specific needs, so that those who can take over their own care will have the necessary tools and those who are more limited will have the appropriate care and services in place to support them. The plans should include an assessment to determine the patient’s capacity to achieve their outlined goals, as well as their knowledge and ability to communicate information about their condition.17
1. Thümler A et al. Clinical characteristics of adults with slowly progressing mucopolysaccharidosis VI: a case series. J Inherit Metab Dis 2012;35(6):1071-1079.
2. Valayannopoulos V et al. Mucopolysaccharidosis VI. Orphanet J Rare Dis 2010;5:5.
3. Swiedler SJ et al. Threshold effect of urinary glycosaminoglycans and the walk test as indicators of disease progression in a survey of subjects with mucopolysaccharidosis VI (Maroteaux–Lamy syndrome). Am J Med Genet A 2005;134A(2):144-150.
4. Giugliani R et al. Management guidelines for mucopolysaccharidosis VI. Pediatrics 2007;120:405-418. 5. Muhlebach MS et al. Respiratory manifestations in mucopolysaccharidoses. Paediatr Respir Rev 2011;12(2):133-138.
6. Lin H-Y et al. Polysomnographic characteristics in patients with mucopolysaccharidoses. Pediatr Pulmonol. 2010;45(12):1205-1212.
7. Kampmann Cet al. Mucopolysaccharidosis VI: cardiac involvement and the impact of enzyme replacement therapy. J Inherit Metab Dis 2014;37(2):269-276.
8. Willoughby CE et al. Anatomy and physiology of the human eye: effects of mucopolysaccharidoses disease on structure and function — a review. Clin Exper Ophthalmol. 2010;38(suppl 1):2-11.
9. Ganesh A et al. An update on ocular involvement in mucopolysaccharidoses. Curr Opin Ophthalmol 2013;24(5):379-388.
10. Kantaputra PN et al. Oral manifestations of 17 patients affected with mucopolysaccharidosis type VI. J Inherit Metab Dis 2014;37(2):263-268.
11.National Institute of Neurological Disorders and Stroke Mucopolysaccharidoses Fact Sheet. http://www.ninds.nih.gov/disorders/mucopolysaccharidoses/detail_mucopolysaccharidoses.htm. Accessed November 2016.
12. Data on file. BioMarin Pharmaceutical Inc.
13. NAGLAZYME [package insert]. Novato, CA: BioMarin Pharmaceutical Inc; 2013.
14. Giugliani R et al. Natural history and galsulfase treatment in mucopolysaccharidosis VI (MPS VI, Maroteaux-Lamy syndrome) — 10-year follow-up of patients who previously participated in an MPS VI Survey Study. Am J Med Genet A 2014;164A(8):1953-1964.
15. Walker R et al. Anaesthesia and airway management in mucopolysaccharidosis. J Inherit Metab Dis 2013;36(2):211-219.
16. Vitale MG et al. Delphi Consensus Report: Best practices in intraoperative neuromonitoring in spine deformity surgery: development of an intraoperative checklist to optimize response. Spine Deformity. 2014;2(5):333-339. 17. American Academy of Pediatrics, American Academy of Family Physicians, American College of Physicians, Transitions Clinical Report Authoring Group, Cooley WC, Sagerman PJ. Supporting the health care transition from adolescence to adulthood in the medical home. Pediatrics 2011;128(1):182-200.
Disease name: Sly | Deficient enzymes: β-glucuronidase | Gene symbol: GUSB
MPS VII, also known as Sly syndrome, is a progressive, multisystemic disease caused by a deficiency of the enzyme β-glucuronidase, which is required for the degradation of the glycosaminoglycans (GAGs) heparan sulphate, dermatan sulphate, and chondroitin sulphate.1
MPS VII can present as early as birth with hydropsis fetalis, or as a young child or adolescent with delayed motor development, mild to severe cognitive impairment and multiorgan disease.1-4
1. Tomatsu S et al. Mutations and polymorphisms in GUSB gene in mucopolysaccharidosis VII (Sly Syndrome). Hum Mutat 2009;30(4):511-519.
2. Gehler J et al. Mucopolysaccharidosis VII: β-glucuronidase deficiency. Humangenetik 1974;23(2):149-158.
3. Bernsen PLJA et al. Phenotypic expression in mucopolysaccharidosis VII. J Neurol Neurosurg Psychiatry 1987;50(6):699-703.
4. Gniadek TJ et al. Cardiovascular pathologies in mucopolysaccharidosis type VII (Sly Syndrome). Cardiovasc Pathol 2015;24(5):322-326.
5. Nampoothiri S et al. Sly disease: mucopolysaccharidosis type VII. Indian Pediatr 2008;45(10):859-861.
6. Yamada Y et al. Treatment of MPS VII (Sly disease) by allogeneic BMT in a female with homozygous A619V mutation. Bone Marrow Transplant 1998;21(6):629-634.
7. Fox JE et al. First human treatment with investigational rhGUS enzyme replacement therapy in an advanced stage MPS VII patient. Mol Genet Metab 2015;114(2):203-208. 8. Muenzer J, Beck M, Eng CM, et al.Genet Med. 2011;13(2):95–101. doi:10.1097/GIM.0b013e3181fea459.
9. Online Mendelian Inheritance in Man, OMIM. Baltimore, MD: Johns Hopkins University Press. http://www.ncbi.nlm.nih.gov/omim. Updated December 20, 2015. Accessed December 21, 2015.
Disease name: Natowicz | Deficient enzymes: Hyaluronidase| Gene symbol: HYAL1
MPS IX, also known at Natowicz syndrome, is caused by a deficiency of the enzyme hyaluronidase, which is required for the degradation of the glycosaminoglycan (GAG) hyaluronan. This GAG is highly expressed in synovial fluid, cartilage, and skin, leading to progressive joint manifestations.1,2
MPS IX can present at varying ages with chronic joint pain unresponsive to anti-inflammatory medication.1,2
1. Imundo L et al. A complete deficiency of Hyaluronoglucosaminidase 1 (HYAL1) presenting as familial juvenile idiopathic arthritis. J Inherit Metab Dis 2011;34(5):1013–1022.
2. Natowicz MR et al. Clinical and biochemical manifestations of hyaluronidase deficiency. N Engl J Med 1996;335(14):1029–1033.
3. Triggs-Raine B et al. Mutations in HYAL1, a member of a tandemly distributed multigene family encoding disparate hyaluronidase activities, cause a newly described lysosomal disorder, mucopolysaccharidosis IX. Proc Natl Acad Sci USA 1999;96(11):6296–6300.