Discover why patients have chosen ROCTAVIAN
How to test for eligibility for ROCTAVIAN?
See the simple steps to identify treatment-eligible patients.
Discover why patients have chosen ROCTAVIAN
Andrew S., dosed with ROCTAVIAN in 2018 as part of a clinical trial.


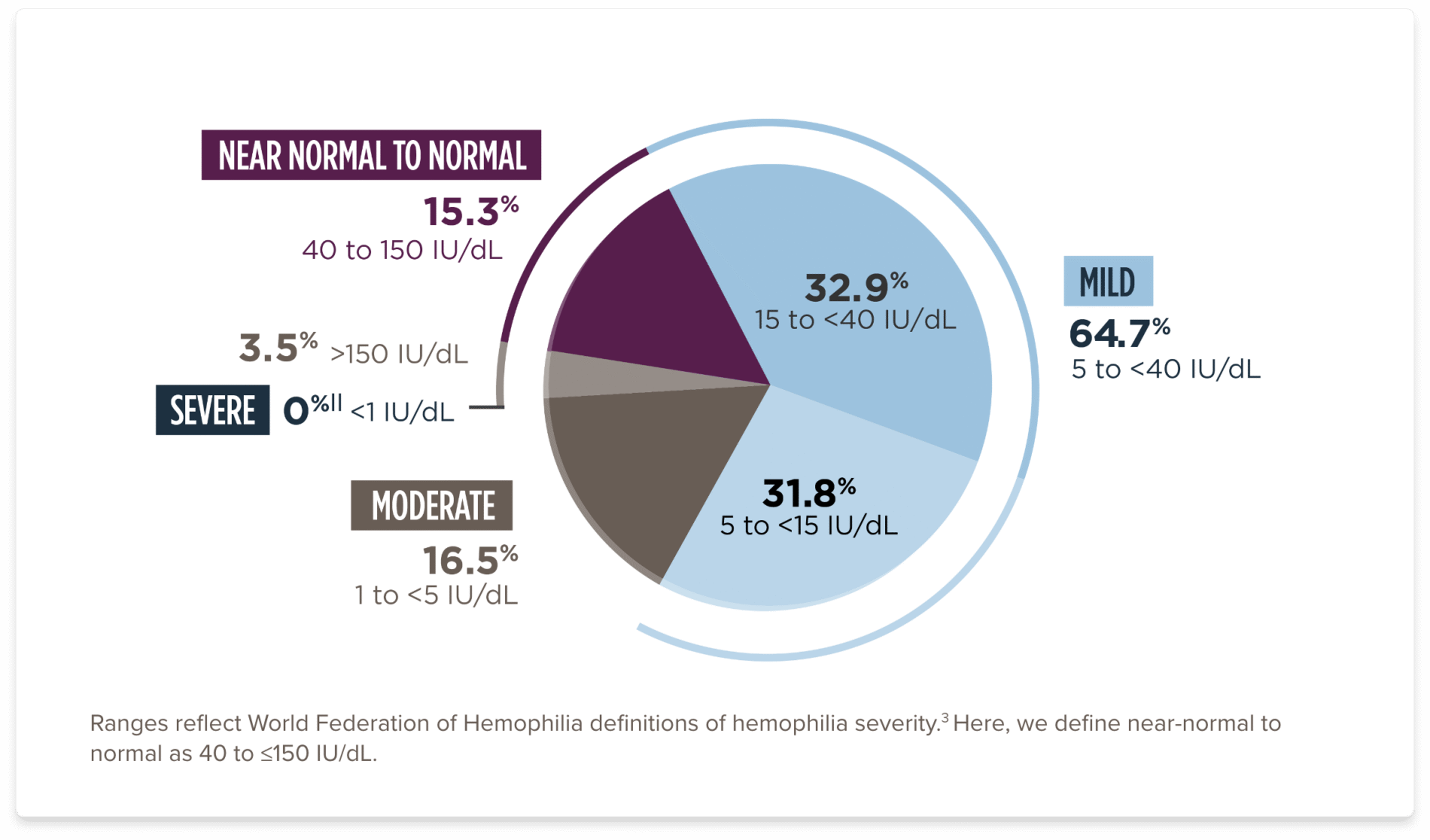
OSA, one-stage assay.
*Median duration of follow-up was 5.0 years (range: 1.7 to 5.2).1
†Of the 134 patients who received ROCTAVIAN in the clinical trial, 112 patients had baseline ABR data prospectively collected during a period of ≥6 months on Factor VIII prophylaxis prior to receiving ROCTAVIAN (rollover population). The remaining 22 patients had baseline ABR collected retrospectively (directly enrolled).2
‡Factor VIII activity produced by ROCTAVIAN in human plasma is higher if measured with OSA compared to chromogenic assay. In clinical trials, there was a high correlation between one-stage and chromogenic Factor VIII activity levels across the entire range of each assay’s results. For routine clinical monitoring of Factor VIII activity levels, either assay may be used. The conversion factor between the assays can be approximated based on clinical trial results (central laboratory) to be: OSA=1.5 X CSA.2
*Median duration of follow-up was 5.0 years (range: 1.7, 5.2).1
†Of the 134 patients who received ROCTAVIAN in the clinical trial, 112 patients had baseline ABR data prospectively collected during a period of ≥6 months on Factor VIII prophylaxis prior to receiving ROCTAVIAN (rollover population). The remaining 22 patients had baseline ABR collected retrospectively (directly enrolled).2
‡Available data from rollover population (n=112) at Week 260.
§Factor VIII activity is not equivalent to response.
||Patients in clinical trial experienced Factor VIII levels of <1%. Under the 5-year analysis of available, valid data, the range was 1.5 to 321.2.
“I would describe my experience with ROCTAVIAN as eye-opening. It’s pretty cool to know that my body is making its own factor now.”
—Andrew S., dosed with ROCTAVIAN in 2018 as part of a clinical trial
The imputed mean ABR (SD) was 5.4 (6.9) at baseline vs 3.9 (7.9) after ROCTAVIAN


*The imputed mean ABR (SD) was 5.4 (6.9) at baseline vs 3.9 (7.9), and the imputed median ABR was 3.3 (range: 0, 34.6) at baseline vs 0.4 (range: 0, 35) after ROCTAVIAN.2
†Median duration of follow-up was 5.0 years (range: 1.7, 5.2).1
‡A noninferiority (NI) test of the difference in ABR during the EEP following administration of ROCTAVIAN was compared with the ABR during the baseline period in the rollover population. The mean difference in ABR was -1.44 (95% CI: -3.28, 0,40) bleeds/year. The NI analysis met the prespecified margin, indicating the effectiveness of ROCTAVIAN.1,2
§Of the 134 patients who received ROCTAVIAN in the clinical trial, 112 patients had baseline ABR data prospectively collected during a period of ≥6 months on Factor VIII prophylaxis prior to receiving ROCTAVIAN (rollover population). The remaining 22 patients had baseline ABR collected retrospectively (directly enrolled).1
‖The 5-year follow-up period started from Study Day 33 (Week 5) or at the end of Factor VIII prophylaxis, including a washout period after treatment with ROCTAVIAN, whichever was later, and ended when a patient completed the study, had the last visit, or withdrew or was lost to follow-up from the study, whichever was earliest.1
¶There were 19 patients (17%) from the 112 rollover population who returned to prophylaxis with median start time at 984 days (range: 33, 1467). An ABR of 35 was imputed for the periods when these patients were on prophylaxis.1




*Median duration of follow-up was 5.0 years (range: 1.7, 5.2).1
†Of the 134 patients who received ROCTAVIAN in the clinical trial, 112 patients had baseline ABR data prospectively collected during a period of ≥6 months on Factor VIII prophylaxis prior to receiving ROCTAVIAN (rollover population). The remaining 22 patients had baseline ABR collected retrospectively (directly enrolled).
§Baseline data were annualized based on a 6-month period of collection.2
‡In the rollover population, a total of 5 patients (4%) did not respond and 27 patients (24%) lost response to treatment with ROCTAVIAN over a median time of 2.7 years (range: 0.96, 4.54). In the directly enrolled population with a longer follow-up, a total of 1 patient (5%) did not respond and 8 patients (36%) lost response to treatment with ROCTAVIAN over a median time of 3.8 years (range: 1.2, 4.8). In the clinical trial, patients who did not respond or lost response returned to prophylaxis.1
The Haemo-QoL-A (Haemophilia-Specific Quality of Life Questionnaire) is designed to measure health-related quality of life specifically for people with hemophilia
Haemo-QoL-A measures included:




EEP=efficacy evaluation period; NI=non-inferiority.
*The EEP started from Week 5 or at the end of Factor VIII prophylaxis including a washout period after treatment with ROCTAVIAN, whichever was later, and ended when a patient completed the study, had the last visit, or withdrew, or was lost to follow-up from the study, whichever was the earliest.
†Of the 134 patients who received ROCTAVIAN in the clinical trial, 112 patients had baseline ABR data prospectively collected during a period of at least 6 months on Factor VIII prophylaxis prior to receiving ROCTAVIAN (rollover population). The remaining 22 patients had baseline ABR collected retrospectively (directly enrolled population).
See the simple steps to identify treatment-eligible patients.
References:
1. Data on file. BioMarin Pharmaceutical Inc.; 2025. 2. ROCTAVIAN® (valoctocogene roxaparvovec-rvox) Prescribing information. BioMarin International, Ltd.; 2023. 3. Srivastava A, Santagostino E, Dougall A, et al. WFH Guidelines for the Management of Hemophilia, 3rd edition. Haemophilia. 2020;26(Suppl 6):1-158. 4. Rentz A, Flood E, Altisent C, et al. Cross-cultural development and psychometric evaluation of a patient-reported health-related quality of life questionnaire for adults with haemophilia. Haemophilia. 2008;14(5):1023-1034. 5. National Institutes of Health. Gene therapy study in severe haemophilia A patients (270-201). Updated December 15, 2023. Accessed August 15, 2025. https://clinicaltrials.gov/study/NCT02576795. 6. Ozelo MC, Mahlangu J, Pasi KJ, et al. Valoctocogene roxaparvovec gene therapy for hemophilia A. N Engl J Med. 2022;386(11):1013-1025. doi:10.1056/NEJMoa2113708
Contraindications: Patients with active infections, either acute (such as acute respiratory infections or acute hepatitis) or uncontrolled chronic (such as chronic active hepatitis B). Patients with known significant hepatic fibrosis (stage 3 or 4 on the Batts-Ludwig scale or equivalent), or cirrhosis, and patients with known hypersensitivity to mannitol.
Infusion-related reactions including hypersensitivity reactions and anaphylaxis, have occurred. Monitor during and for at least 3 hours after ROCTAVIAN administration. Administer ROCTAVIAN in a setting where personnel and equipment are immediately available to treat infusion-related reactions. Discontinue infusion for anaphylaxis.
Hepatotoxicity: The safety and effectiveness of ROCTAVIAN in patients with hepatic impairment has not been established. Perform liver health assessments prior to administration. The majority of patients treated with ROCTAVIAN experienced ALT elevations and required corticosteroids for ALT elevation. Assess patient’s ability to receive corticosteroids and/or other immunosuppressive therapy that may be required for an extended period. Live vaccines should not be administered to patients while on immunosuppressive therapy.
Monitor ALT weekly for at least 26 weeks and as clinically indicated, during corticosteroid therapy and institute corticosteroid treatment in response to ALT elevations as required. Continue to monitor ALT until it returns to baseline. Monitor Factor VIII activity levels since ALT elevation may be accompanied by a decrease in Factor VIII activity. One case of autoimmune hepatitis was reported during third year follow-up in a patient with history of hepatitis C and steatohepatitis.
It is recommended that patients abstain from consuming alcohol for at least 1 year after administration and thereafter limit alcohol use. Concomitant medications may cause hepatotoxicity, decrease Factor VIII activity, or change plasma corticosteroid levels which may impact liver enzyme elevation and/or Factor VIII activity or decrease the efficacy of the corticosteroid regimen or increase their side effects. Closely monitor concomitant medication use including herbal products and nutritional supplements and consider alternative medications in case of potential drug interactions.
Thromboembolic Events: Factor VIII activity above ULN has been reported following ROCTAVIAN infusion. Thromboembolic events may occur in the setting of elevated Factor VIII activity above ULN. Evaluate patients for risk of thrombosis including general cardiovascular risk factors before and after administration of ROCTAVIAN. Advise patients on their individual risk of thrombosis in relation to their Factor VIII activity levels above ULN and consider prophylactic anticoagulation. Advise patients to seek immediate medical attention for signs or symptoms indicative of a thrombotic event.
Factor VIII Inhibitors and Monitoring for Inhibitors. The safety and effectiveness of ROCTAVIAN in patients with prior or active Factor VIII inhibitors have not been established. Patients with active Factor VIII inhibitors should not take ROCTAVIAN. Following administration, monitor patients for Factor VIII inhibitors (neutralizing antibodies to Factor VIII). Test for Factor VIII inhibitors especially if bleeding is not controlled, or plasma Factor VIII activity levels decrease.
Monitor Factor VIII using the same schedule for ALT monitoring. It may take several weeks after ROCTAVIAN infusion before ROCTAVIAN-derived Factor VIII activity rises to a level sufficient for prevention of spontaneous bleeding episodes. Exogenous Factor VIII or other hemostatic products may also be required in case of surgery, invasive procedures, trauma, or bleeds. Consider more frequent monitoring in patients with Factor VIII activity levels ≤5 IU/dL and evidence of bleeding, taking into account the stability of Factor VIII levels since the previous measurement.
Factor VIII activity produced by ROCTAVIAN in human plasma is higher if measured with one-stage clotting assays compared to chromogenic substrate assays. When switching from hemostatic products prior to ROCTAVIAN treatment, physicians should refer to the relevant prescribing information to avoid the potential for Factor VIII activity assay interference during the transition period.
Malignancy: The integration of liver-targeting AAV vector DNA into the genome may carry the theoretical risk of hepatocellular carcinoma development. ROCTAVIAN can also insert into the DNA of other human body cells. Monitor patients with risk factors for hepatocellular carcinoma (eg, hepatitis B or C, nonalcoholic fatty liver disease, chronic alcohol consumption, nonalcoholic steatohepatitis, advanced age) with regular liver ultrasound (eg, annually) and alpha-fetoprotein testing for 5 years following ROCTAVIAN administration. In the event that any malignancy occurs after treatment with ROCTAVIAN, contact BioMarin Pharmaceutical Inc. at 1-866-906-6100.
Most Common Adverse Reactions: Most common adverse reactions (incidence ≥5%) were nausea, fatigue, headache, infusion-related reactions, vomiting, and abdominal pain. Most common laboratory abnormalities (incidence ≥10%) were ALT, AST, LDH, CPK, Factor VIII activity levels, GGT, and bilirubin >ULN. Patients also experienced adverse reactions from corticosteroid use.
Isotretinoin, Efavirenz, and HIV-Positive Patients. Isotretinoin is not recommended in patients who are benefiting from ROCTAVIAN. Efavirenz is not recommended in patients treated with ROCTAVIAN. Clinical studies of ROCTAVIAN did not include sufficient numbers of patients with HIV to determine whether the efficacy and safety differs compared to patients without HIV infection.
Females and Males of Reproductive Potential. ROCTAVIAN is not intended for administration in women. There are no data on the use of ROCTAVIAN in pregnant women or regarding lactation. For 6 months after administration of ROCTAVIAN, men of reproductive potential and their female partners must prevent or postpone pregnancy using an effective form of contraception, and men must not donate semen.
You may report side effects to the FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. You may also report side effects to BioMarin Pharmaceutical Inc. at 1-866-906-6100.
Please see additional safety information in the Prescribing Information.
Indication
ROCTAVIAN® (valoctocogene roxaparvovec-rvox) is indicated for the treatment of adults with severe hemophilia A (congenital Factor VIII deficiency with Factor VIII activity <1 IU/dL) without antibodies to adeno-associated virus serotype 5 detected by an FDA-approved test.